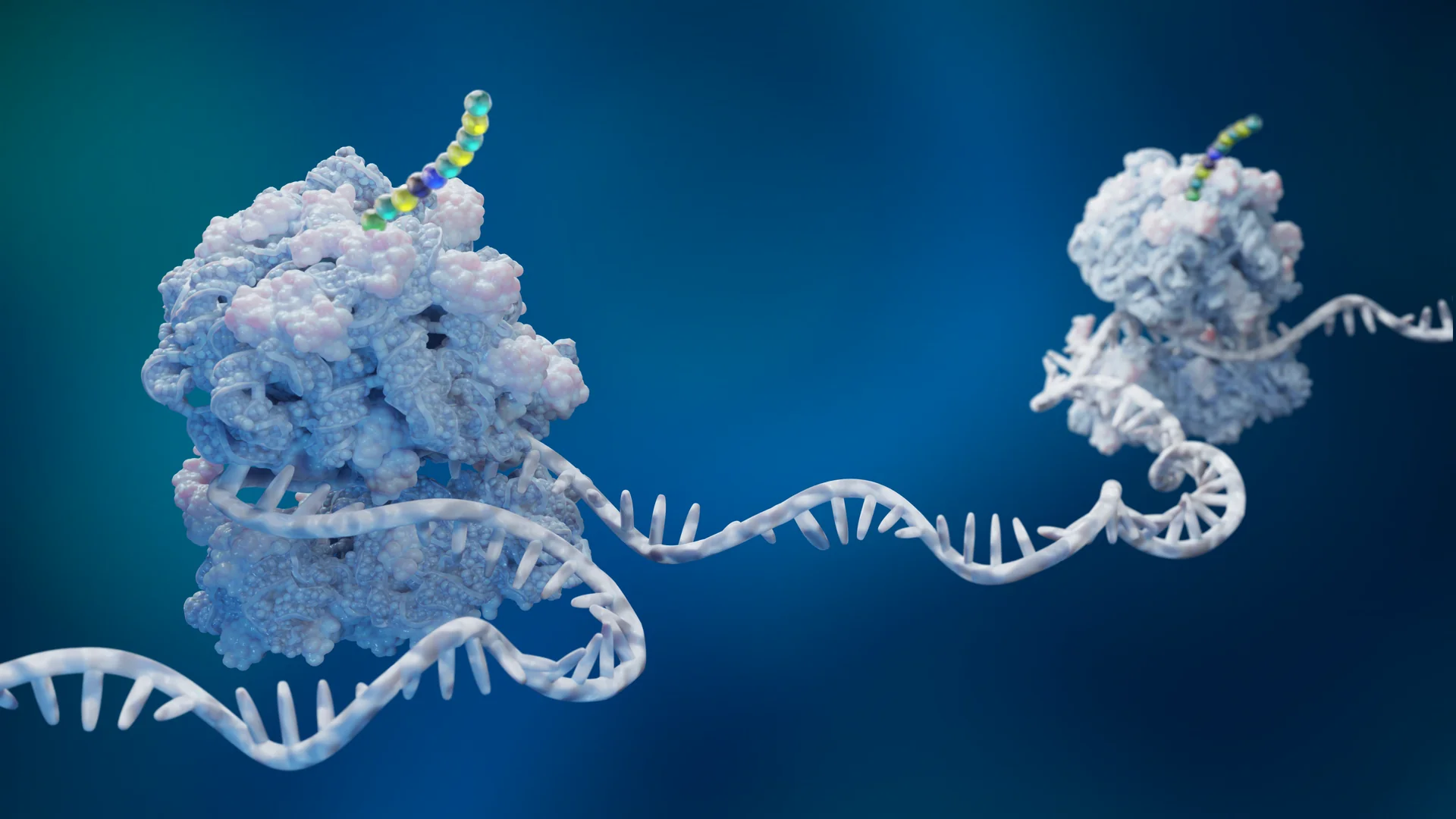
Engineering the future of biology with revolutionary genome synthesis and expanded genetic code technology.
Enhanced ADC linkers
-
ncAA
Various -
Incorporation molecule
Trastuzumab -
Impact
Enhanced ADC linkers
Description
Antibody–drug conjugates (ADCs) are a fast-growing class of targeted cancer therapeutics. They combine the precision of monoclonal antibodies with the power of small-molecule cytotoxics. Most approved ADCs rely on the same linker: the protease-cleavable valine–citrulline (Val–Cit) dipeptide. It was designed to release payloads after cleavage by lysosomal cathepsins. In practice, this is a challenge. Val–Cit is cut by proteases in healthy tissues. Payload leaks early. Toxicity follows.
By incorporating ncAAs, the authors of this work identified peptide linkers with high selectivity for cancer-associated proteases. This overcomes the core limitation of conventional linker design. The outcome is clear: potent tumor cell killing; minimal activity from non-cleavable controls and clean separation of on-target versus off-target effects!
Citation: Gorzeń et al., 2025
Most approved antibody-drug conjugates use valine-citrulline (Val-Cit) linkers that are cleaved by lysosomal cathepsins. The problem: these proteases also exist in healthy tissues, causing premature payload release and off-target toxicity. Better linker selectivity requires chemistry beyond the standard amino acid toolkit.
Using non-canonical amino acid residues in the linker peptide, researchers identified sequences with high selectivity for cancer-associated proteases over normal-tissue enzymes. The ncAA-containing linkers achieved potent tumour cell killing while non-cleavable controls showed minimal activity, confirming that therapeutic effect depends on selective cleavage, not passive drug leakage (Gorzen et al., 2025).
Linker design is a critical variable in ADC development. Site-specific ncAA conjugation (defining where the drug attaches to the antibody) combined with ncAA-enhanced linkers (controlling when and where the drug releases) gives developers two independent optimisation levers for improving the therapeutic window of antibody-drug conjugates.